Primer Schemes
About
Both artic tools and the artic pipeline are designed to work with tiling amplicon schemes for targeted enrichment of viral genomes. The protocol is described in the Quick J et al. 2017 Nature Protocols paper and the schemes are produced by Primal Scheme.
Tiling amplicon schemes contain primers that are designed using a greedy algorithm against multiple reference genomes. Tiling refers to the fact that neighbouring amplicons overlap one another, ensuring that complete coverage of the target genome is acheived. To prevent short overlap products being produced between neighbouring amplicons, 2 primer pools are used to alternate primer pairs. The following figure from the Quick J et al. 2017 paper shows how primers from 2 pools are used to tile across a reference genome:
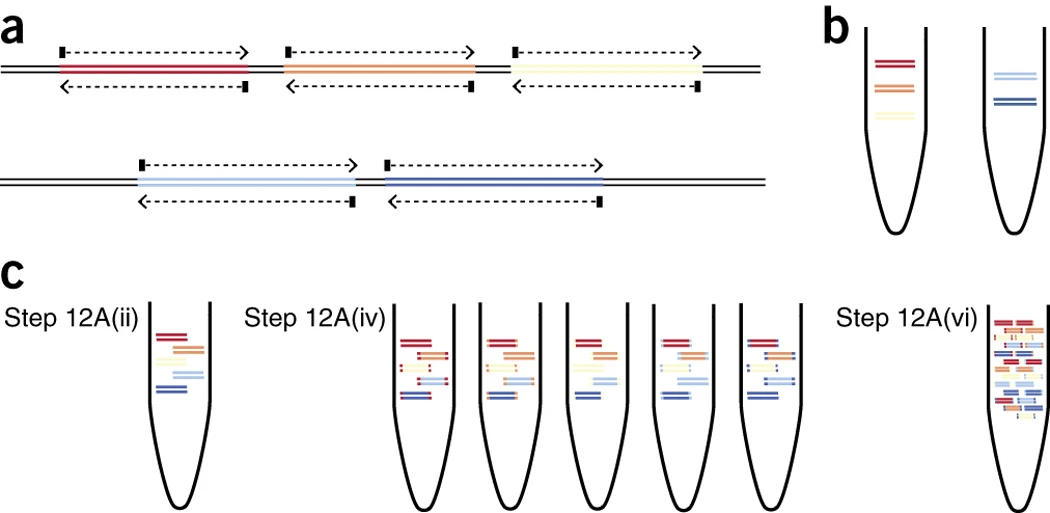
Quick J et al. 2017 (a) Schematic showing the regions amplified in pools 1 (upper track) and 2 (lower track), and the intended overlap between pools (as determined in Step 1)...
Availability
Frequently used primer schemes for viral genome sequencing are found in the ARTIC primer scheme repo. These include Nipah, Ebola and SARS-CoV-2 primer schemes. Multiple scheme versions may be available, with a higher version number denoting schemes that have had improvements made to them (e.g. introduction of alternate primers).
ARTIC primer schemes can be downloaded using the artic-tools get_scheme command. For example:
artic-tools get_scheme ebola --schemeVersion 2
The artic pipeline will also attempt to download a scheme using artic tools if a scheme can not be found locally.
File format
Primer schemes produced by Primal Scheme come with several files (which you can read more about here). The 2 files which are needed for artic are:
| file extension | description |
|---|---|
*.reference.fasta |
the sequence of the reference genome used in the scheme |
*.primer.bed |
the coordinates of each primer in the scheme, relative to the reference genome |
Note: *.primer.bed was formerly *.scheme.bed, but has lately changed to reflect a slightly updated file format. The old *.scheme.bed should still work fine and is available alongside the new versions at the ARTIC primer scheme repo.
The *.primer.bed file is in 6-column BED format, with the following column descriptions:
| column | name | type | description |
|---|---|---|---|
| 1 | chrom | string | primer reference sequence |
| 2 | chromStart | int | starting position of the primer in the reference sequence |
| 3 | chomEnd | int | ending position of the primer in the reference sequence |
| 4 | name | string | primer name |
| 5 | primerPool | int | primer pool* |
| 6 | strand | string | primer direction (+/-) |
* column 5 in the BED spec is an int for score, whereas here we are using it to denote primerPool.
Note: BED format is a 0-based, half-open format. This means that reference sequence position counting starts at 0 and the chromEnd is not included in the primer sequence.
Scheme logic
The following sections will discuss how primer schemes are read from a *.primer.bed file and used in the ARTIC software.
Reading schemes
primers
Each line in a *.primer.bed file is a single primer. Lines are processed one at a time and the input file does not need to be sorted in any way beforehand (although most schemes are sorted by primer start coordinate). As lines are read, they are converted to primer objects.
Most of the logic behind creating a primer object relies on the primer name (column 4). As a primer line is read, we check for the following tags in the name field:
| tag | meaning |
|---|---|
_LEFT |
the left primer |
_RIGHT |
the right primer |
_alt |
the primer is an alt |
Important:
- tags are case sensitive
- tags can be anywhere in the primer name but typically are placed at the end (e.g. REGION_42_RIGHT_alt)
- a
_LEFTor_RIGHTtag is required and only one is allowed per primer - the
_alttag is optional and denotes that the primer is an alternate version of another primer
To merge a primer with its alternate into a single primer object, the two primers are combined such that a maximal span is achieved. The merged primer will have _alt dropped from the ID.
Depending on the presence of the _LEFT or _RIGHT tag, the primer is assigned a direction (+/- to denote forward/reverse). Direction is also encoded in column 6 of newer schemes.
In addition to the information encoded in the name field, we also use the start and end coordinates (columns 2 and 3) and the primer pool (column 5) when we create primer objects. The primer pool is encoded as an integer in the later scheme versions, but we treat it as a string in the primer scheme code (for backwards compatibility with the older primer scheme files that stored it as a string).
For each primer object, the following information is available:
- primer name
- primer base name (all tags removed)
- reference start coordinate
- reference end coordinate
- number of alts merged into the primer
- primer pool
We can also call several helper methods on the object for getting things such as primer sequence, length etc.
schemes
Once all lines in a *.primer.bed file have been read and converted to primer objects they are held in a scheme. In artic-tools, this consists of 2 unordered maps, one for forward primers and one for reverse primers, as well as some additional datastructures to allow validation, index and query of schemes.
Validating schemes
Note: the following sections apply to
artic-tools
On reading the scheme file, it will have satisified the following checks:
- scheme file must exist and be readable
- scheme file must have at least 5 columns (tab separated)
- each row must encode a primer (problem rows are flagged and validation fails after all rows are tried)
- scheme file must not contain multiple reference sequence IDs (first column)
The scheme data structure will then be validated:
- the scheme must contain primers
- the number of forward primers must match the number of reverse primers (this is after merging alts)
- each forward primer must have a reverse primer with a matching base name within the same primer pool
- no gaps must be present within the scheme (i.e. regions of the reference not covered)
Querying schemes
Once loaded and validated, the primer scheme can be queried for basic information such as:
- number of primers
- number of primer pools
- reference sequence name
- reference region covered by the scheme
For a given a reference coordinate it, the scheme can also return the nearest forward and reverse primer. The returned pair of primers is provided within a container, referred to as an amplicon.
amplicons
Any primers within a scheme can be combined to produce an amplicon if they satisfy the following:
- one primer is forward and one is reverse
- the end coordinate of the forward primer is before the start coordinate of the reverse primer (i.e. primers can't be outward facing of each other)
The scheme will arrange any amplicon so the forward primer within an amplicon is first. It will then mark the the amplicon as properly paired if the following conditions are met:
- the base name of the forward and reverse primer match
- the primer pool name matches
Amplicons are just containers for primers, so don't hold information directly. But, as well being able to access the contained primers, the following information can be inferred:
- pool name ('no pool' given if amplicon not properly paired)
- amplicon name (combines the forward and reverse primer names)
- maximum amplicon span (includes primer sequence)
- minimum amplicon span (excludes primer sequence)
The primer scheme will pre-compute expected amplicons and store the mean span etc.
finding closest primers given a position in the reference
To find a the closest primers to a given reference coordinate, the primer scheme builds the following in addition to the maps of forward and reverse primers:
- sorted vector of start coordinates for each forward primer in the scheme
- sorted vector of end coordinates for each reverse primer in the scheme
Both of these are vectors of pairs, where the first is the coordinate and the second is a key to lookup the corresponding primer in the map.
Pseudocode for primer search:
// X = sorted vector of start coordinates and primers for forward primers
// Y = sorted vector of end coordinates and primers for reverse primers
function findClosestPrimer(pos, coordinates) {
// get the first coordinate that is >= pos
primer = lower_bound(coordinates.begin(), coordinates.end(), pos)
// check coordinate is closer to pos than next smallest coordinate
if abs(primer->first - pos) <= abs(primer.previous()->first - pos)
return primer->second // primer name
else
return primer.previous()->second // primer name
}
function findPrimerPair(pos) {
fPrimer = findClosestPrimer(pos, X)
rPrimer = findClosestPrimer(pos, Y)
return amplicon(fPrimer, rPrimer) // return an amplicon
}
check position containment in amplicon overlaps or primer sequences
To check if a position is contained by multiple amplicons, or by a primer sequence, the primer scheme precomputes a list of locations to check against.
To do this a bit vector is used to record presence/absence at each reference position. For recording amplicon overlap, every position covered by >1 amplicon has a bit set to 1. Another bit vector can be used to store all the locations where primers from a given pool are located etc.
In practice a single bit vector is used where the bit vector length is equivalent to the reference genome size, multiplied by the number of containment checks offered by the scheme. The number of checks is used to offset the bit vector access. The bit vectors are initialised when the primer scheme is validated.
Pseudocode for containment checks:
// X = sorted vector of start coordinates and primers for forward primers
// Y = sorted vector of end coordinates and primers for reverse primers
// L = length of reference sequence
// BV = the empty bit vector used to record positions
// setBits is a helper function to set bits in a contiguous region of a bit vector
function setBits(start, end) {
while start != end {
BV[start] = 1
start++
}
}
// createIndex sets up a bit vector with containment locations
function createIndex() {
// initalise the bit vector to fit amplicon overlaps
// and primer locations for pools 1 and 2
BV = [0] * (L * 3)
// get the amplicon overlap locations
for i = 0; i < len(X)-1; i++ {
if X[i+1] < Y[i] {
setBits(Y[i], X[i+1])
}
}
// get the primer sequence locations
for primer in X and Y {
if primer in pool1 {
setBits((primer.start + (L * 1)), (primer.end + (L * 1)))
}
if primer in pool2 {
setBits((primer.start + (L * 2)), (primer.end + (L * 2)))
}
}
}
// queryContainment queries the bit vector
function queryContainment(pos, type) {
if type == ampliconOverlap
return BV[pos + (L * 0)]
if type == primerPool1
return BV[pos + (L * 1)]
if type == primerPool2
return BV[pos + (L * 2)]
}